
Indications for Testing
A two year old male child born to a non-consanguineous couple (30 yrs, male and 29 yrs, female) was referred for genetic counseling in view of the child having the symptoms of febrile and non febrile myoclonic seizures and developmental delay.
Birth and Development History:
- Development was reported to be normal until 6 months of age. At 6 months of age he had a febrile myoclonic seizure. Neuroregression was reported after 6 months.
- He has had 9 episodes of febrile myoclonic seizure and one episode ofa non-febrile myoclonic seizure. He is currently on anti-epileptic drugs.
Genetic Counseling/Family History
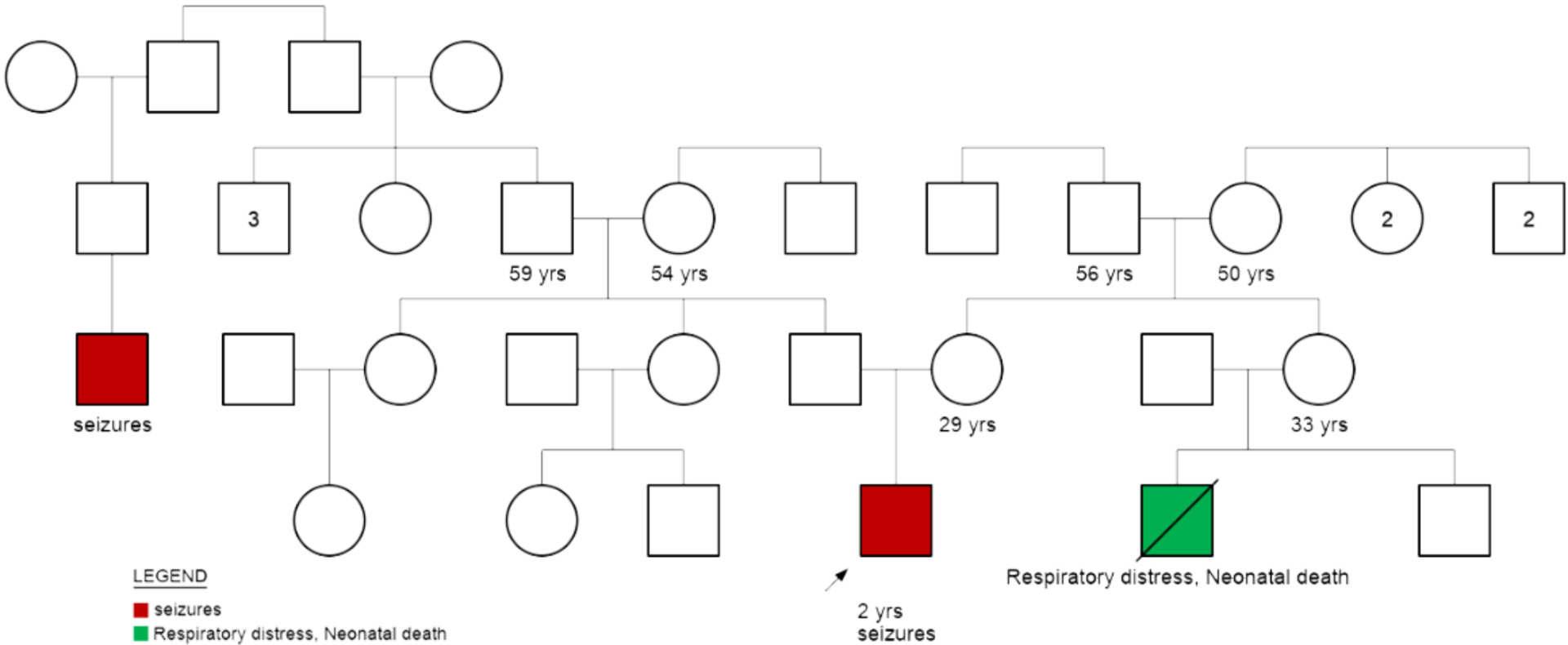
Previous genetic testing: Whole exome sequencing was performed at another laboratory facility which was reported to be normal. The genetic tests most utilized in the evaluation of children with epilepsy include chromosomal microarray (CMA), epilepsy gene panels, and whole-exome sequencing (WES). Each test has its own specific benefits and limitations, and the utility of different tests may vary for a given individual. WES may identify genetic mutations in approximately 30% of patients with epilepsy. CMA may be abnormal in approximately 10% of individuals with epilepsy and is more likely to be abnormal in children with epilepsy and other conditions such as autism, developmental delay, and/or intellectual disability.
Genetic Finding
- CMA picked up a heterozygous, pathogenic deletion spanning about 1.6MB on chromosome 2 at 2q24.3.
- The heterozygous deletion is associated with Migrating epilepsy or Atypical Dravet syndrome
- Genes involved in this deletion include: CSRNP3, GALNT3, TTC21B, SCN1A, SCN9A, SCN7A and XIRP2.
2q24.3 microdeletion
2q24.3 microdeletion is a rare deletion that presents a frequency of incidence < 1/1000000 and involves both female and male sexes. 2q24.3 microdeletion can present with a variety of phenotypes. The most likely features are seizures, psychiatric diseases, developmental delay, learning difficulty or disability, severe growth failure after birth (possibly with low birth weight), some unusual facial characteristics, and cardiac involvement.
The 2q24.2-24.3 chromosome region encodes the cluster of sodium channel genes, which are important in severe childhood epilepsy phenotypes such as Dravet syndrome and migrating partial seizures of infancy. Dravet syndrome is a treatment-resistant developmental epileptic encephalopathy that is characterized by multiple types of seizures including bouts of debilitating status epilepticus. SCN1A variants are a frequent cause of Dravet Syndrome.
Recommendations
Parental testing: To rule out if parents are carriers, chromosomal microarray and karyotype were advised for mother and father. Parents' reports were normal.
- For future pregnancies: CMA may be offered in the future pregnancy to rule out any chromosomal abnormalities. In rare cases, parents may be carriers of germline mosaicism.
Conclusion
NGS based tests such as clinical exome, whole exome or a panel of genes is often opted for epilepsy. This case illustrates that in cases where the panel NGS is negative, CMA may be beneficial to look for CNV changes as it may be associated with epilepsy.


